WHAT WE DO
Together, the Patrick and Watson labs study the interplay between intracellular bacterial pathogens and the innate immune cells that sense and respond to them.
The Watson lab is focused on the notorious human pathogen, Mycobacterium tuberculosis, which remains a major global health threat. M. tuberculosis has evolved a variety of specific adaptations to not only survive but also replicate within the harsh environment inside a macrophage. We want to understand how host macrophages sense and respond to M. tuberculosis and how specific host proteins regulate the outcome of M. tuberculosis infection, both inside macrophages ex vivo and in a mouse model of infection. We have recently started to focus on the interface between mitochondria and M. tuberculosis. Specifically, we are interested in uncovering the molecular mechanisms that explain why mutations in several genes related to mitochondrial function confer susceptibility to M. tuberculosis in humans.
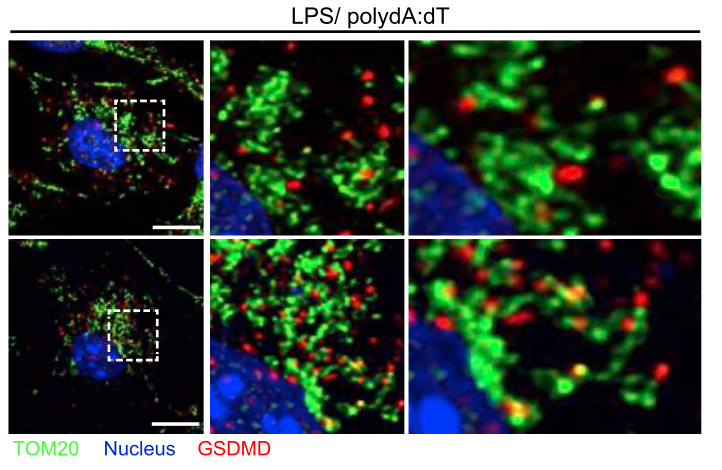
Localization of the pore-forming protein gasdermin D to the mitochondrial network following inflammasome activation in primary murine macrophages (from Weindel et al.., 2022, Cell)
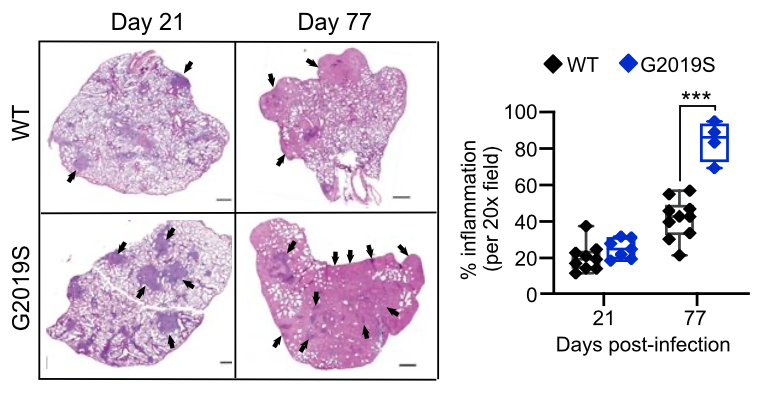
Mice harboring the LRRK2 G2019S mutation are susceptible to hyperinflammation during M. tuberculosis infection (from Weindel et al., 2022, Cell)
The Patrick lab is passionate about post-transcriptional regulation of gene expression in eukaryotic cells. Using a multi-disciplinary toolset, we probe the molecular mechanisms that macrophages use to activate an innate immune response that is rapid, robust, and regulated. We mainly study how RNA binding proteins control the ability of macrophages to respond to infection, using a variety of bacterial and viral models. By working to uncover how RNA binding proteins work and how macrophages functionalize RNA binding proteins to orchestrate a fine-tuned innate immune response, our work furthers our understanding of a variety of human diseases. We have also started to study how myeloid cancer-associated splicing factor mutations alter inflammation and innate immune responses.
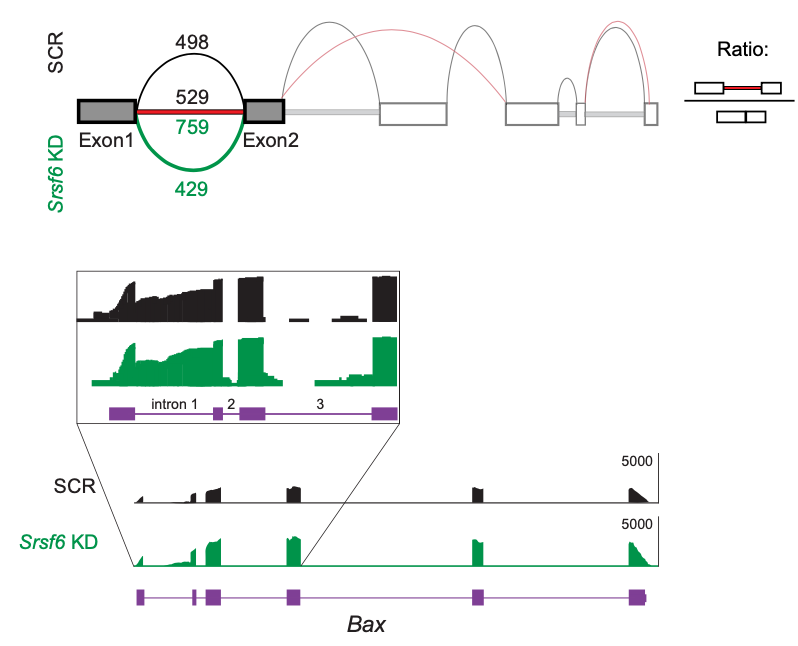
The splicing factor SRSF6 is required to maintain mitochondrial homeostasis via controlling the alternative splicing of the apoptosis protein BAX (from Wagner et al., 2022, Elife)

RNA binding proteins in the SR and hnRNP families shape the innate immune transcriptome in murine macrophages in distinct and profound ways (from Wagner, Scott, West et al, 2021. Front Immunol.)